









조로증 또는 허친슨-길포드 증후군
조로증은 어린이의 조기 노화를 특징으로 하는 희귀 유전 질환입니다.
조로증의 정의
허친슨-길포드 증후군으로도 알려진 조로증은 희귀 유전 질환입니다. 신체의 노화가 증가하는 것이 특징입니다. 이 병리를 가진 아이들은 노년기의 초기 징후를 보입니다.
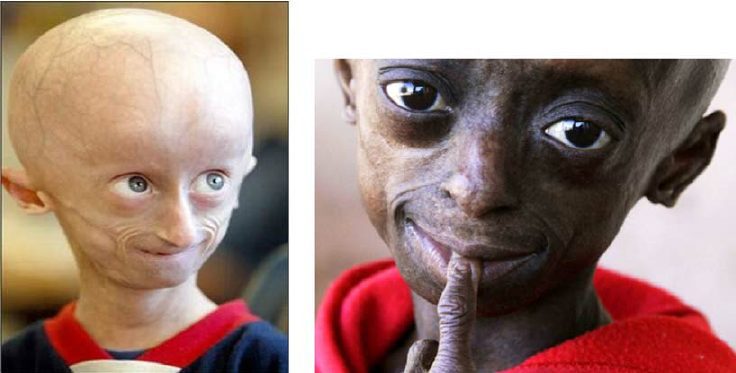
태어날 때 아이는 아무런 이상을 나타내지 않습니다. 유아기까지는 "정상"으로 나타납니다. 점차적으로 그의 형태와 신체가 비정상적으로 발달합니다. 그는 정상보다 덜 빨리 자라며 또래의 체중이 늘지 않습니다. 면에서 개발도 지연되고 있다. 그는 두드러진 눈(안도에서, 강하게 전진), 매우 가늘고 구부러진 코, 얇은 입술, 작은 턱 및 튀어나온 귀를 나타냅니다. Progeria는 또한 심각한 탈모(탈모증), 노화된 피부, 관절 결핍 또는 심지어 피하 지방량(피하 지방)의 손실의 원인입니다.
아동의 인지 및 지적 발달은 일반적으로 영향을 받지 않습니다. 그것은 본질적으로 운동 발달에 대한 결핍 및 결과이며, 앉거나 서거나 심지어 걷는 데 어려움을 유발합니다.
Hutchinson-Gilford 증후군 환자는 또한 일부 동맥이 좁아져 죽상동맥경화증(동맥 막힘)이 발생합니다. 그러나 동맥경화증은 심장마비 또는 심지어 뇌혈관 사고(뇌졸중)의 위험을 증가시키는 주된 요인입니다.
이 질병의 유병률(전체 인구 중 영향을 받는 사람의 수)은 전 세계적으로 1/4백만 신생아에 해당합니다. 따라서 희귀병입니다.
조로증은 상염색체 우성 유전 질환이기 때문에 그러한 질병이 발병할 위험이 가장 큰 개체는 부모에게도 조로증이 있는 사람입니다. 무작위 유전자 돌연변이의 위험도 가능합니다. Progeria는 질병이 가족 서클에 존재하지 않더라도 모든 개인에게 영향을 줄 수 있습니다.
조로증의 원인
조로증은 희귀 유전 질환입니다. 이 병리학의 기원은 LMNA 유전자 내의 돌연변이 때문입니다. 이 유전자는 단백질 형성을 담당합니다: 라민 A. 후자는 세포 핵 형성에 중요한 역할을 합니다. 그것은 핵막(세포의 핵을 둘러싸고 있는 막)의 형성에 필수적인 요소입니다.
이 유전자의 유전적 돌연변이로 인해 라민 A가 비정상적으로 형성됩니다. 이 비정상적으로 형성된 단백질은 세포핵의 불안정성과 유기체 세포의 조기 사멸의 원인이 됩니다.
과학자들은 현재 이 단백질이 인간 유기체의 발달에 관여하는 것에 대한 자세한 정보를 얻기 위해 이 주제에 대해 스스로에게 묻고 있습니다.
Hutchinson-Gilford 증후군의 유전적 전달은 상염색체 우성 유전을 통해 발생합니다. 특정 유전자의 두 복사본 중 하나만(어머니 또는 아버지로부터) 전달되면 질병이 아이에게 발병하기에 충분합니다.
또한, LMNA 유전자의 무작위 돌연변이(부모 유전자의 전달로 인한 것이 아님)도 이러한 질병의 기원일 수 있습니다.
조로증의 증상
조로증의 일반적인 증상은 다음과 같은 특징이 있습니다.
- 신체의 조기 노화 (어린 시절부터);
- 정상보다 체중 증가가 적습니다.
- 아이의 작은 크기;
- 안면 기형: 가늘고 구부러진 코, 돌출된 눈, 작은 턱, 돌출된 귀 등 ;
- 서거나 앉거나 걷는 데 어려움을 일으키는 운동 지연;
- 동맥경화증의 주요 위험 인자인 동맥의 협착.
조로증의 위험 요소
조로증은 드문 유전적 유전성 상염색체 우성 질환이므로 두 부모 중 한 사람에게 질병이 있는 것이 가장 중요한 위험 요소입니다.
조로증에 대한 치료법은 무엇입니까?
조로증과 관련된 증상은 진행성이며 환자의 사망으로 이어질 수 있습니다.
현재 이 질병에 대한 치료법은 없습니다. 조로증의 유일한 관리는 증상의 관리입니다.
그런 다음 그러한 질병을 치료할 수 있는 치료법을 찾기 위한 연구가 각광을 받고 있습니다.